
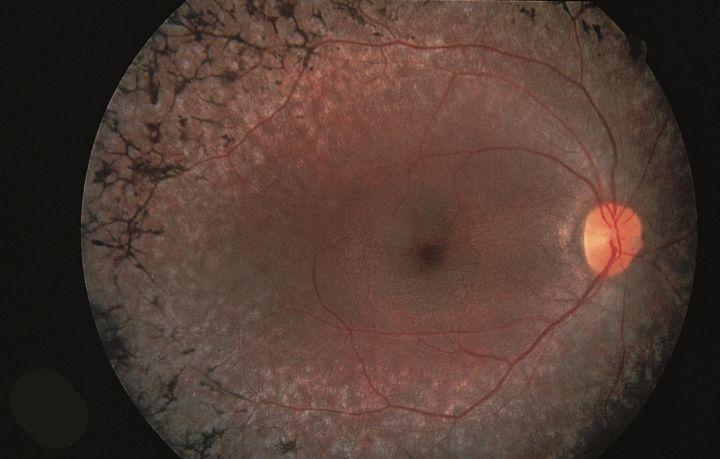
Konference iedzimtu tīklenes distrofiju un citu reto acu slimību pacientiem, vecākiem un speciālistiem
2023. gada 2. februārī norisinājās tiešsaistes konference iedzimtu tīklenes distrofiju un citu reto acu slimību pacientiem, pacientu vecākiem, ārstiem, redzes un cita veida funkcionāliem speciālistiem - "Pacienti, profesionāļi, slimība – kopā mēs varam vairāk".

